


Autor: Dinler Antunes, pós-doutorando no Departamento de Ciências da Computação da Rice University, no Texas/EUA
A apresentação de peptídeos endógenos na superfície celular representa uma rota central para a resposta imunológica adaptativa. Este mecanismo, presente em praticamente todas as células nucleadas do organismo, possibilita que linfócitos T circulantes (CD8+) "fiscalizem" o conteúdo intracelular destas células apresentadoras, permitindo a detecção de infecções virais e tumores. Esta rota pode ser explicada de maneira simples, sendo visivelmente uma espécie de "controle de qualidade" do conteúdo intracelular (Figura 1). Os detalhes moleculares desta rota, no entanto, são bastante complexos e variáveis. Por exemplo, se tentarmos entender quais são os requisitos responsáveis pela capacidade de um peptídeo de desencadear a ativação de um linfócito, perceberemos o envolvimento de múltiplas variáveis relacionadas a diferentes etapas deste processo. Em primeiro lugar, o peptídeo precisa ser capaz de "sobreviver" ao processamento, uma vez que nem todos os possíveis peptídeos contidos em uma dada proteína são apresentados na superfície (Yewdell et al., 2003). Um ponto importante nesta etapa de processamento diz respeito a capacidade de ligação dos peptídeos gerados às moléculas de MHC de classe I (sigla em inglês para Complexo Principal de Histocompatibilidade). Hoje temos acesso a uma ampla coleção de dados sobre ligação de peptídeos a moléculas de MHC e é possível inclusive predizer a afinidade de ligação do complexo considerando-se as sequências das moléculas envolvidas (Backert & Kohlbacher, 2015; Karosiene et al., 2012). No final desta rota, o conjunto peptídeo:MHC (pMHC) poderá interagir com linfócitos circulantes e o desfecho da resposta dependerá de uma série de fatores envolvidos na afinidade da interação entre o pMHC e o receptor de linfócito T (ou TCR, na sigla em inglês). Em um capítulo de livro publicado em 2012 nós utilizamos o termo reatividade para nos referirmos a esta etapa final e os fatores envolvidos na ativação dos linfócitos T citotóxicos. No mesmo capítulo, nós discutimos a importância de um "nível de seleção" intermediário entre o processamento e a reatividade, relativo a estabilidade do complexo pMHC.
Embora intuitivamente possamos imaginar que a estabilidade de um dado pMHC será um reflexo da afinidade de ligação do complexo, de modo que apenas pMHCs estáveis serão formados no retículo endoplasmático e consequentemente apresentados na superfície, esta relação nem sempre é observada. Na verdade, os dados sugerem que a estabilidade do pMHC é inclusive um melhor preditor da imunogenicidade do peptídeo do que a própria afinidade de ligação (van der Burg at al., 1996; Harndahl et al., 2012; Jorgensen et al. 2014). Embora os já conhecidos "resíduos âncoras" sejam primariamente envolvidos na afinidade do complexo, as cadeias laterais de outros resíduos secundários podem influenciar positivamente ou negativamente na manutenção do modo de ligação mais favorável, o que pode levar a instabilidade do complexo (Jorgensen et al. 2014). Isso acaba tendo impacto direto na possível estimulação de linfócitos, sobretudo considerando que uma diversidade de peptídeos está sendo apresentada ao mesmo tempo e que estes diferentes complexos pMHC estão competindo pela interação com o TCR; complexos mais numerosos e mais estáveis tem maior chance de serem reconhecidos (dependendo também dos fatores envolvidos na reatividade).
O servidor netMHCstab utiliza redes neurais para predizer a estabilidade de um dado complexo pMHC com base na análise de sequencias, tendo apresentado resultados promissores na identificação de epitopos de linfócito T quando combinado a um método consenso para predição de afinidade de ligação ao MHC (Jorgensen et al. 2014). No entanto, estes métodos baseados em análise de sequência são muito dependentes da qualidade e do volume de dados utilizado para treinar as predições (Wang et al., 2014). Esta restrição muitas vezes impede a generalização das predições para outros alelos de MHC ou outros tamanhos de ligantes, visto que os dados experimentais ainda são limitados para a maioria dos alelos (considerando-se apenas os MHCs de classe I clássicos, são mais de 10 mil alelos descritos em humanos). Métodos estruturais tem potencial para permitirem uma maior generalização das predições, especialmente considerando-se uma característica dinâmica como estabilidade. Em tese, tendo-se uma estrutura inicial do complexo é possível calcular as forças envolvidas na interação e simular o comportamento do complexo em solução. Durante seu trabalho de Doutorado desenvolvido na UFRGS, meu colega Maurício Menegatti Rigo utilizou métodos de docking e dinâmica molecular para estudar a estabilidade de ligantes na fenda do MHC. Utilizando dados obtidos de simulações considerando a real estrutura das moléculas envolvidas (full-atom MD) foi possível observar claros sinais de instabilidade em peptídeos mutados (Figura 2), além de identificar algumas interações que parecem ser mais importantes para a manutenção do modo de ligação.
A dinâmica molecular é a ferramenta de bioinformática estrutural com a maior precisão em termos da correta descrição das forças envolvidas e das trajetórias moleculares preditas para um dado sistema. No entanto, esta precisão acarreta um elevado custo computacional que em muitos casos inviabiliza a análise de sistemas muito grandes, ou a observação de eventos que ocorrem em escalas de tempo superiores a centenas de microssegundos. O trabalho descrito acima corrobora o potencial preditivo da dinâmica molecular quanto a instabilidade do complexo, mas nestas simulações curtas (20 ns) não é possível observar o completo "desprendimento" do ligante. Na verdade, um trabalho recente demonstrou que o desprendimento completo pode não ser observado mesmo em uma dinâmica molecular de 1000 ns (1 µs), a mais longa simulação de um complexo pMHC já publicada (full-atom MD) (Knapp et al., 2015). Para conseguir observar este evento em nível molecular, os autores implementaram uma estratégia alternativa. Em primeiro lugar, ao invés de considerar a descrição atômica das moléculas envolvidas, eles utilizaram uma representação simplificada dos resíduos (coarse-grained 3-point model). Em segundo lugar, eles utilizaram um método de segmentação hierárquica da proteína para restringir a flexibilidade de grupos de resíduos, o que também melhora a performance (reduz graus de liberdade) e previne a perda do correto enovelamento da proteína (unfolding). Os movimentos neste sistema foram então explorados através do método de Monte Carlo. Finalmente, eles realizaram repetidas etapas de simulated annealing para permitir uma rápida exploração do espaço conformacional sem ficar restrito a mínimos locais de energia. Esta abordagem permitiu observar o completo desprendimento da fenda em um tempo computacionalmente viável (Figura 3) e foi capaz de discriminar entre ligantes e não ligantes (AROC = 0,85). Esta é a primeira vez em que são descritas conformações intermediárias do ligante durante o processo de desprendimento do MHC, possibilitando novos insights e revelando outros detalhes moleculares importantes para o desencadeamento de uma resposta imunológica celular. A capacidade de predizer dados experimentais corrobora a hipótese de que as simplificações realizadas não prejudicaram a correta descrição das moléculas simuladas, mas futuros experimentos serão necessários para determinar a real acurácia e a potencial generalização dos resultados observados.
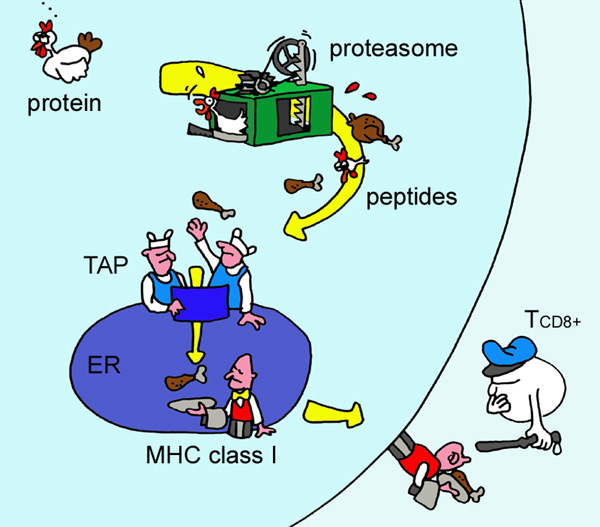 |
Figura 1. Ilustração de Eric Reits para representar a rota
de apresentação de peptídeos endógenos. Uma amostra das proteínas intracelulares é processada e apresentada na superfície celular no contexto do MHC-I, permitindo seu reconhecimento pelos linfócitos T (Yewdell et al., 2003). |
Embora intuitivamente possamos imaginar que a estabilidade de um dado pMHC será um reflexo da afinidade de ligação do complexo, de modo que apenas pMHCs estáveis serão formados no retículo endoplasmático e consequentemente apresentados na superfície, esta relação nem sempre é observada. Na verdade, os dados sugerem que a estabilidade do pMHC é inclusive um melhor preditor da imunogenicidade do peptídeo do que a própria afinidade de ligação (van der Burg at al., 1996; Harndahl et al., 2012; Jorgensen et al. 2014). Embora os já conhecidos "resíduos âncoras" sejam primariamente envolvidos na afinidade do complexo, as cadeias laterais de outros resíduos secundários podem influenciar positivamente ou negativamente na manutenção do modo de ligação mais favorável, o que pode levar a instabilidade do complexo (Jorgensen et al. 2014). Isso acaba tendo impacto direto na possível estimulação de linfócitos, sobretudo considerando que uma diversidade de peptídeos está sendo apresentada ao mesmo tempo e que estes diferentes complexos pMHC estão competindo pela interação com o TCR; complexos mais numerosos e mais estáveis tem maior chance de serem reconhecidos (dependendo também dos fatores envolvidos na reatividade).
 |
| Figura 2. Visualização dos dados obtidos de uma dinâmica molecular de um complexo pMHC não estável. Os cones verdes indicam a direção e a intensidade das forças observadas em determinados resíduos. Neste exemplo, elas indicam uma forte tendência de desprendimento da porção C-terminal do ligante. Estes dados diferem daqueles observados para um ligante controle. Modificado da Tese de Doutorado de Maurício M. Rigo (2015). |
A dinâmica molecular é a ferramenta de bioinformática estrutural com a maior precisão em termos da correta descrição das forças envolvidas e das trajetórias moleculares preditas para um dado sistema. No entanto, esta precisão acarreta um elevado custo computacional que em muitos casos inviabiliza a análise de sistemas muito grandes, ou a observação de eventos que ocorrem em escalas de tempo superiores a centenas de microssegundos. O trabalho descrito acima corrobora o potencial preditivo da dinâmica molecular quanto a instabilidade do complexo, mas nestas simulações curtas (20 ns) não é possível observar o completo "desprendimento" do ligante. Na verdade, um trabalho recente demonstrou que o desprendimento completo pode não ser observado mesmo em uma dinâmica molecular de 1000 ns (1 µs), a mais longa simulação de um complexo pMHC já publicada (full-atom MD) (Knapp et al., 2015). Para conseguir observar este evento em nível molecular, os autores implementaram uma estratégia alternativa. Em primeiro lugar, ao invés de considerar a descrição atômica das moléculas envolvidas, eles utilizaram uma representação simplificada dos resíduos (coarse-grained 3-point model). Em segundo lugar, eles utilizaram um método de segmentação hierárquica da proteína para restringir a flexibilidade de grupos de resíduos, o que também melhora a performance (reduz graus de liberdade) e previne a perda do correto enovelamento da proteína (unfolding). Os movimentos neste sistema foram então explorados através do método de Monte Carlo. Finalmente, eles realizaram repetidas etapas de simulated annealing para permitir uma rápida exploração do espaço conformacional sem ficar restrito a mínimos locais de energia. Esta abordagem permitiu observar o completo desprendimento da fenda em um tempo computacionalmente viável (Figura 3) e foi capaz de discriminar entre ligantes e não ligantes (AROC = 0,85). Esta é a primeira vez em que são descritas conformações intermediárias do ligante durante o processo de desprendimento do MHC, possibilitando novos insights e revelando outros detalhes moleculares importantes para o desencadeamento de uma resposta imunológica celular. A capacidade de predizer dados experimentais corrobora a hipótese de que as simplificações realizadas não prejudicaram a correta descrição das moléculas simuladas, mas futuros experimentos serão necessários para determinar a real acurácia e a potencial generalização dos resultados observados.
 |
| Figura 3. Simulação do processo de desprendimento de um peptídeo da fenda do MHC utilizando (A) uma nova metodologia baseada em coarse-grained Monte Carlo e simulated annealing (HNMMC) ou (B) dinâmica molecular. O peptídeo utilizado (AAAKTPVIV) foi previamente descrito como não ligante de HLA-A*0201, alelo utilizado nestes experimentos. A técnica HNMMC foi capaz de descrever um completo desprendimento da fenda, em um tempo muito inferior ao da dinâmica molecular (C). Figura publicada em Knapp et al., 2015. |

